{kind=link}
GLP1-ELP and ELP-FGF21 cotreatment has potent weight-reducing effects
To test our hypothesis that GLP-1 and FGF21 act additively—or better—to control glycemia and inhibit weight gain, we carried out a short-term pilot study comparing a GLP-1/FGF21 combination therapy to each respective single-drug treatment. This pilot study used long-acting analogs of GLP-1 and FGF21, each fused to an ELP, that we previously developed and validated for the treatment of T2D (17, 18). ELPs are repetitive peptide polymers characterized by a (VPGXaaG)n sequence, where “Xaa” is any amino acid besides proline and “n” is the number of repeats (19, 20). A notable feature of ELPs is their reversible lower critical solution temperature (LCST) phase behavior in aqueous medium (21). ELPs have a distinct and tunable “transition temperature” (Tt)—also referred to as the cloud point temperature. Below their Tt, ELPs are miscible in water, and above their Tt, they form a water-immiscible coacervate (21), and this thermal responsiveness is retained when an ELP is genetically fused to a peptide or protein drug (22). By manipulating the Tt—via choice of the Xaa residue and the molecular weight (23)—an ELP-drug fusion can be designed to form a depot under the skin that steadily releases molecules into systemic circulation (17, 18, 24). GLP1-ELP and ELP-FGF21 fusions are active in vitro and form subcutaneous depots capable of blood glucose–lowering effects for at least 5 days in diabetic mice following a single injection (17, 18).
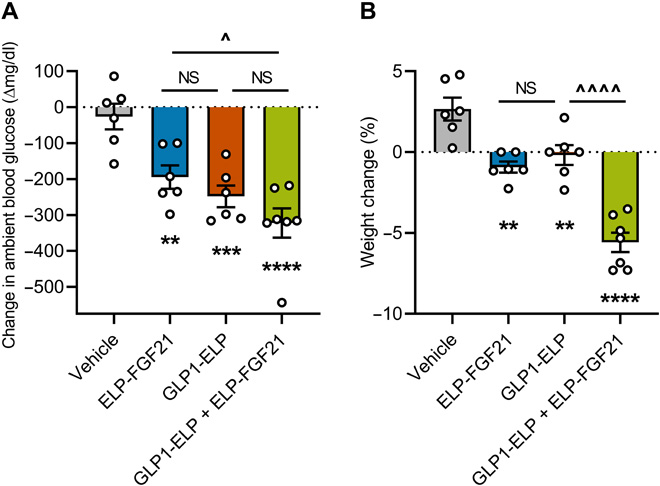
Db/db mice were injected subcutaneously with GLP1-ELP, ELP-FGF21, an equimolar mixture of GLP1-ELP and ELP-FGF21, or vehicle control. Ambient blood glucose levels and body weights were measured 48 hours after injection and reported as a change from preinjection baseline. All treatments significantly reduced blood glucose levels compared to vehicle, while combination treatment resulted in blood glucose levels that trended even lower than each respective single drug (Fig. 1A). Treatment with ELP-FGF21 or GLP1-ELP effectively inhibited weight gain (−0. 9 ± 0.3 and −0.2 ± 0.6%) compared to vehicle-treated mice, which gained 2.7 ± 0.7% body weight in 48 hours (Fig. 1B). In contrast, mice treated with the combination of ELP-FGF21 and GLP1-ELP exhibited a robust 5.6 ± 0.6% reduction in body weight (Fig. 1B). Together, these data suggest that GLP-1 and FGF21 act at least additively to induce weight loss and possibly to improve glycemic control in diabetic mice, motivating the development of a modular drug with GLP-1 and FGF21 dual agonism while retaining the ELP component for sustained release.

Six-week-old db/db mice (n = 6 to 7) were subcutaneously injected with ELP-FGF21 (1000 nmol/kg), GLP1-ELP (1000 nmol/kg), or GLP1-ELP and ELP-FGF21 (1000 nmol/kg each). Ambient blood glucose levels (A) and body weights (B) were measured 48 hours after injection and reported as a magnitude change from pretreatment baseline and a percentage change from preinjection weight. Data are presented as means ± SEM and were analyzed by one-way ANOVA, followed by Tukey’s tests. *, treatment compared to vehicle; ^, comparisons between treatments; ^P < 005, **P < 0.01, ***P < 0.001, and ****/^^^^P < 0.0001. NS, not significant (P > 0.05).
Design and production of a unimolecular GLP-1/FGF21 dual agonist
The dual agonist was designed as a head-to-tail polypeptide fusion protein, with GLP-1 located at the N terminus, FGF21 at the C terminus, and an intervening ELP (“GLP1-ELP-FGF21”). This orientation provided a solvent-exposed N terminus for GLP-1, and an exposed C terminus for FGF21, both of which are essential to activate their respective receptors (6, 25), while the linear architecture enabled facile synthesis and scale-up in a bacterial expression system. The ELP served a dual role as both a flexible linker—providing sufficient physical separation between the two drugs to enable dual receptor binding—and a module to create an injectable depot and thereby enable sustained release of the drug from the injection site.
GLP1-ELP-FGF21 used mutations in FGF21 to promote protein stability (18) and in GLP-1 to stabilize the α helix and protect the N terminus from proteolytic cleavage (24). GLP-1 also incorporated a di-alanine (AA) leader that facilitated recombinant expression, but that is removed in vivo by dipeptidyl peptidase 4 (DPP4) to expose an active N terminus (24). The ELP sequence was strategically chosen on the basis of its Tt, and it consisted of (VPGXG)120 with a 4:1 valine:alanine ratio in the Xaa residue position (see table S1 for the complete amino acid sequence). The GLP1-ELP-FGF21 fusion protein was recombinantly expressed in Escherichia coli and was purified from crude cell lysate by inverse transition cycling (ITC), a nonchromatographic method developed by our research group that exploits that LCST phase transition behavior imparted to the ELP fusion by the ELP tag (22, 26). A 72-kDa band associated with the full-length GLP1-ELP-FGF21 fusion product was visible in SDS–polyacrylamide gel electrophoresis (PAGE) throughout the purification process (Fig. 2A), and ITC purification alone was sufficient to isolate the dual agonist from contaminants.

(A) SDS-PAGE analysis of the 72-kDa fusion protein following recombinant expression in E. coli and ITC-based purification. 1, molecular weight ladder (kilodaltons); 2, cell lysate; 3, insoluble lysate fraction; 4, soluble lysate fraction; 5, hot spin supernatant; 6 to 8, ITC rounds 1 to 3. (B and C) In vitro GLP-1 and FGF21 activity assays for GLP1-ELP-FGF21. GLP-1R agonism (B) was measured by quantifying adenosine 3′,5′-monophosphate (cAMP) production in human embryonic kidney (HEK) 293 cells stably expressing the GLP-1R and the cAMP-inducible luciferase reporter. Cells were stimulated for 5 hours with GLP1-ELP-FGF21, the GLP1-ELP single-agonist control, or native GLP-1. FGF21 receptor agonism (C) was measured by quantifying extracellular signal–regulated kinase 1/2 (ERK1/2) phosphorylation in HEK293 cells stably expressing FGF receptor 1 (FGFR1) and β-klotho and by normalizing phospho-ERK1/2 to total ERK1/2. Cells were stimulated for 5 min with GLP1-ELP-FGF21, the ELP-FGF21 single-agonist control, or native FGF21. Data are presented as means ± SEM, n = 3. (D to F) LCST phase transition behavior of GLP1-ELP-FGF21. (D) The optical density (OD) at 350 nm of GLP1-ELP-FGF21 at the indicated concentration in phosphate-buffered saline (PBS) was measured as a function of temperature, with temperature ramped at a rate of 1°C/min. (E) Turbidity versus temperature scans were repeated as in (D) for the indicated concentrations (n = 3). Tt values were defined as the temperature corresponding to the 50% maximum OD and are plotted as a function of fusion protein concentration. The horizontal dashed line indicates the approximate temperature of the subcutaneous space in a mouse (27). (F) A turbidity scan was repeated for GLP1-ELP-FGF21 at an injection-relevant concentration (150 μM), with the temperature ramped up to 37°C and then down to 20°C.
GLP1-ELP-FGF21 has dual agonism at the GLP-1 and FGF21 receptors
The half-maximal effective concentration (EC50) for the GLP-1 and FGF21 dual-agonist components were measured using in vitro activity assays in cells stably expressing either the GLP-1R or the FGF21 receptor complex. Fusion of GLP-1 to an ELP increased the GLP-1R EC50 approximately 10-fold (Fig. 2B), which agrees with our previous findings (17), while the GLP-1R EC50 of GLP1-ELP-FGF21 (23.9 ± 5.7 pM) was not different from that of GLP1-ELP (29.5 ± 5.0 pM) (Fig. 2B). Fusion of FGF21 to an ELP increased the FGF21 receptor EC50 approximately 20-fold (Fig. 2C), which is also consistent with our previous reports (18). The dual agonist had a marginally greater FGF21 receptor EC50 (43.2 ± 8.4 nM) compared to ELP-FGF21 (18.8 ± 4.5 nM) (Fig. 2C), but this difference was not statistically significant (P > 0.05). Together, these data demonstrate that simultaneous presentation of GLP-1 and FGF21 on an ELP does not significantly affect the activity of each drug.
Phase transition behavior of GLP1-ELP-FGF21 is suitable for depot formation
The LCST phase transition behavior of the GLP1-ELP-FGF21 fusion was evaluated by monitoring the optical density (OD) of a solution of the fusion protein as a function of temperature, defining the Tt as the temperature at which the solution becomes turbid. On the basis of previous optimization studies by our group (17, 18), we have identified a target Tt range between 27° and 32°C as suitable for depot formation—triggered by body heat—with ELP-drug release kinetics that are appropriate for once-weekly dosing. An ELP fusion with a Tt of <27°C forms an excessively stable coacervate that exhibits poor drug absorption, while a fusion with a Tt near 35°C [the temperature of the mouse subcutaneous space (27)] exhibits a bolus-like release profile (17). GLP1-ELP-FGF21 was confirmed to have LCST phase change behavior, with a Tt between 27° and 29°C at the injection-relevant concentration range of 100 to 200 μM (Fig. 2D). The Tt was concentration dependent (Fig. 2E), and the dual-agonist phase change behavior was reversible (Fig. 2F). The reversibility and inverse dependence of Tt on fusion protein concentration (28) are attributes critical to the controlled release capabilities of ELP-based drug depots: As fusion protein molecules at the depot margin are diluted, their Tt rises above body temperature, thereby reversing the LCST phase transition and allowing the release of ELP-drug fusion molecules from the depot.
A GLP1-ELP-FGF21 dual-agonist fusion protein has sustained dose-dependent effects on body weight and glycemia
The dual-agonist drug was next tested for efficacy in diabetic mice. The db/db mouse model was selected because of its extreme degree of hyperglycemia paired with obesity, as high baseline body weight and glycemic levels provided a large window to identify additive effects of dual agonism. Mice were administered either a single subcutaneous injection of GLP1-ELP-FGF21 at the indicated dose or vehicle. Ambient blood glucose levels were measured daily until all cohorts returned to baseline levels. Significant reductions in blood glucose versus time area under the curve (AUC) were observed at the two highest doses tested (750 and 1000 nmol/kg) (Fig. 3A); however, raw blood glucose versus time data revealed that it was not effect size but rather effect duration that increased in a dose-dependent manner (Fig. 3B). All doses reduced blood glucose levels from >300 to <150 mg/dl (Fig. 3B), with effects persisting for 4 days at the lowest dose (250 nmol/kg) and 8 days at the highest dose tested (1000 nmol/kg) (Fig. 3B). This dose-response trend aligns with previous observations (17) and is consistent with an ELP-based depot platform that releases drug at a steady rate for a duration proportional to initial dosing size. Compared to vehicle control, all tested doses of GLP1-ELP-FGF21 had an inhibitory effect on weight gain, while the two highest doses tested induced weight loss—reducing body weights by up to 7.2 ± 2.3% (Fig. 3C). On day 7 after treatment, a net weight loss effect persisted in the groups (750 and 1000 nmol/kg) (−2.6 ± 3.3 and −2.7 ± 3.7%, respectively) (Fig. 3D).

(A to D) Six-week-old db/db mice (n = 3 to 4) received a single subcutaneous injection of GLP1-ELP-FGF21 at the indicated dose or vehicle. Ambient blood glucose levels were measured every 24 hours until animals returned to baseline levels and are reported as blood glucose versus time AUC or raw values (A and B). Body weights were recorded daily and are reported as a percentage change from preinjection weight over time (C) or on day 7 after injection (D). (E) GLP1-ELP-FGF21 (1000 nmol/kg) was subcutaneously administered to 6-week-old db/db mice (n = 4) as a radiolabeled protein, blood samples were collected at indicated time points following injection, and plasma gamma counts were correlated to fusion protein concentration. Regression curves were fit to the terminal portion of the dataset, and data could be described by either a first-order (dotted) or a zero-order (solid) elimination model. Data are presented as means ± SEM and were analyzed by one-way ANOVA, followed by Dunnett’s tests. *P < 0.05 and **P < 0.01.
To confirm that sustained efficacy was the result of prolonged drug in circulation—as is observed when an ELP depot forms at the site of injection (17, 18)—plasma drug levels were measured over time following a single subcutaneous injection of GLP1-ELP-FGF21 to db/db mice. Aside from a modest burst release in the first 24 hours, plasma drug levels remained steady near 100 nM out to day 10 (Fig. 3E), at which point drug levels decreased at a rate consistent with first-order elimination (29). When a linear regression curve was fit to the terminal portion of the data, the absorption half-life was calculated to be 7.6 ± 1.1 days (table S2). The data were described nearly as well by a zero-order elimination model (R2 = 0.81) as by a first-order model (R2 = 0.85) (Fig. 3E and table S2). The likely zero-order release kinetics observed here are consistent with ELP-based drug depots observed previously (17) and correspond to the release of fusion molecules into circulation at a constant rate. When pharmacokinetic data were analyzed alongside the blood glucose–versus–time efficacy data (1000 nmol/kg) (Fig. 3B), 100 nM appeared to be the minimal therapeutic concentration, as blood glucose levels returned to baseline in the same time frame that serum drug levels dropped below 100 nM (on or after day 10).
In summary, treatment of obese and hyperglycemic mice with a GLP-1/FGF21 dual-agonist drug had potent and sustained effects on body weights and ambient blood glucose levels. A single injection was sufficient to maintain therapeutic drug levels and protect mice from hyperglycemia and weight gain for >7 days, demonstrating the suitability of a GLP1-ELP-FGF21 depot for a once-weekly dosing scheme. A dose of 1000 nmol/kg was identified as yielding a maximal therapeutic effect over the intended dosing cycle and was selected for further evaluation.
A GLP1-ELP-FGF21 dual agonist outperforms a long-acting GLP-1RA and a GLP-1/FGF21 drug mixture in diabetic mice
To elucidate the relative contribution of GLP-1 and FGF21 to the potent in vivo effects of the dual agonist, as well as understand the impact of incorporating both drugs into a single molecule, we next compared the efficacy of GLP1-ELP-FGF21 with that of a GLP1-ELP single-agonist monotherapy, an ELP-FGF21 monotherapy, and an equimolar mixture of GLP1-ELP and ELP-FGF21. To ensure consistency across treatments, the same ELP sequence was used in each drug formulation. The previously characterized ELP-FGF21 fusion (18) incorporates an ELP of equal length and composition to that comprising the dual agonist (table S1), however, a fusion of GLP-1 to the same ELP was not available from previous studies; hence, we produced the relevant GLP1-ELP fusion (table S1). To do so, a vector encoding GLP-1 fused at its C terminus to the ELP of interest [(VPGXG)120 with a 4:1 valine:alanine ratio at the Xaa residue position] was expressed in E. coli and purified by ITC as previously described (17). A 52-kDa band associated with the GLP1-ELP fusion was visible by SDS-PAGE following purification (fig. S1A). The appropriately designed GLP1-ELP fusion was tested for GLP-1R agonism and exhibited the anticipated 10-fold increase in EC50 compared to native GLP-1 (fig. S1B).
The LCST phase transition behavior was evaluated for GLP1-ELP and the equimolar mixture of GLP1-ELP and ELP-FGF21, revealing Tt values of 27.5°and 28°C, respectively, at the injection-relevant concentration of 200 μM (fig. S1, C and D). The Tt values were concentration dependent (fig. S1, E and F), and the phase behavior of each fusion/fusion mixture was reversible (fig. S1, G and H). Note that the LCST phase transition behavior of ELP-FGF21 has been characterized previously (18).
Next, pharmacokinetic profiles were evaluated for the synthesized GLP1-ELP fusion, as well as the equimolar mixture of GLP1-ELP and ELP-FGF21. Both GLP1-ELP and the mixture exhibited steady plasma drug levels consistent with sustained release from a subcutaneous depot (Fig. 4A and fig. S1I). The GLP1-ELP monotherapy depot behaved similarly to that of the dual agonist, releasing fusion molecules into circulation at a rate that could be described by the zero-order elimination model (table S2) and resulting in plasma drug levels hovering near 100 nM for at least 10 days (fig. S1I). The 1:1 mixed depot released GLP1-ELP and ELP-FGF21 fusion molecules at different rates (Fig. 4A). The ELP-FGF21 component reached a higher maximum serum concentration (Cmax) than the GLP1-ELP component (table S2), and ELP-FGF21 plasma levels dropped below 100 nM by day 4, while GLP1-ELP plasma levels hovered steadily around 100 nM for 7 days (Fig. 4A). Although absorption of each component of the mixture fit the zero-order release model (table S2), neither GLP1-ELP nor ELP-FGF21 maintained steady plasma levels for as long as GLP1-ELP-FGF21 did, suggesting a pharmacokinetic advantage to delivering the two drugs in a unimolecular format. Note that sustained release of ELP-FGF21 from a subcutaneous depot has been previously reported (18).

(A) A mixed ELP depot releases GLP-1 and FGF21 at different rates. Six-week-old db/db mice (n = 4 to 5) received a single subcutaneous injection consisting of a 1:1 mixture of the synthesized GLP1-ELP and previously reported ELP-FGF21 (18). The mixture was tested once when GLP1-ELP was radiolabeled and once when ELP-FGF21 was radiolabeled. Fusions were injected at 200 μM and dosed at 1000 nmol/kg each of GLP1-ELP and ELP-FGF21. Blood samples were collected at indicated time points following injection, and plasma gamma counts were measured and correlated to fusion protein concentration. Lines represent regression curves fit to the terminal portion of each dataset. Data can be described by both the first-order (dotted) and zero-order (solid) elimination models. (B to G) Six-week-old db/db mice (n = 6 to 7) were treated weekly for 4 weeks with GLP1-ELP-FGF21, GLP1-ELP, ELP-FGF21, or PBS vehicle. Fusion proteins were administered subcutaneously at 1000 nmol/kg. (B to D) Glucose challenge. Seventy-two hours after the first treatment cycle, mice were fasted 5 hours, baseline blood glucose levels were measured (B), and mice were injected intraperitoneally with glucose (0.75 g/kg). Blood glucose levels were measured at indicated time points (C), and blood glucose versus time AUC values were calculated (D). (E and F) Body weights and food consumption were measured every 1 to 2 days and are reported as percentage change from preinjection weights (E) and cumulative food intake per mouse (F). (G) Before the first treatment (day 0) and 6 days following the final treatment (day 27), percent glycated hemoglobin A1c (%HbA1c) was measured in all cohorts and reported as a magnitude change from prestudy values. Data are presented as means ± SEM and were analyzed by one-way ANOVA or two-way repeated-measures ANOVA, followed by Dunnett’s tests. *P < 0.05, ***P < 0.001, and ****P < 0.0001.
After establishing appropriate controls, we compared the acute and chronic metabolic effects of dual-agonist treatment to that of the respective single-agonist ELP fusion proteins. Db/db mice were treated weekly for 4 weeks with GLP1-ELP-FGF21 (1000 nmol/kg), GLP1-ELP (1000 nmol/kg), ELP-FGF21 (1000 nmol/kg), or vehicle. An intraperitoneal glucose tolerance test (GTT) (0.75 mg/kg) was performed 3 days after the first dosing cycle, when body weights were consistent across treatment groups (fig. S2A) and each drug was at a maximally therapeutic plasma concentration (Fig. 3E and fig. S1I) (18). Fasting blood glucose levels measured before the GTT revealed a stepwise decrease following treatment with ELP-FGF21, GLP1-ELP, and GLP1-ELP-FGF21, respectively, with levels trending lowest in the dual-agonist cohort (125 ± 6.0 mg/dl) (Fig. 4B). All treatments improved glucose tolerance compared to vehicle (Fig. 4C) and were further analyzed by AUC (Fig. 4D), which sets a consistent baseline of 0 mg/dl due to the varying t = 0 blood glucose levels for each respective cohort. ELP-FGF21 monotherapy treatment significantly decreased GTT AUC compared to vehicle, while GLP1-ELP monotherapy significantly decreased AUC compared to ELP-FGF21 (Fig. 4D). Meanwhile, GLP1-ELP-FGF21 treatment resulted in the lowest AUC (Fig. 4D) and was the only cohort to recover to near-baseline levels within 60 min (Fig. 4C), indicating a degree of glucose tolerance afforded only when FGF21 and GLP-1 are administered in combination.
Chronic dosing with the GLP1-ELP monotherapy significantly inhibited weight gain over the 4-week treatment period compared to vehicle (Fig. 4E). The reduced rate of weight gain in this cohort was likely due in part to a significant reduction in food intake (Fig. 4F), which is consistent with the anorectic effect associated with GLP-1RA therapy. Dual-agonist treatment further reduced weight gain compared to GLP1-ELP monotherapy over the 4-week study (+14.1 ± 1.6% versus +25.3 ± 1.9%, respectively) (Fig. 4E), despite GLP1-ELP-FGF21–treated mice consuming chow at an equivalent rate to GLP1-ELP–treated mice (Fig. 4F). Thus, the weight reduction observed in the dual-agonist cohort could not be attributed exclusively to an anorectic effect and likely incorporated an additional mechanism of action involving energy expenditure. Note that ELP-FGF21 monotherapy did not induce a significant effect on body weight (Fig. 4E), indicating that cooperative action between FGF21 and GLP-1 is necessary for realizing the full weight loss effect.
Percent glycated hemoglobin A1c (%HbA1c) was measured before the initiation of treatment (day 0) and at the termination (day 27) of the chronic dosing study. %HbA1c reflects mean circulating glucose levels over time and only shifts with red blood cell turnover (30). The average life span for a red blood cell is ~40 days in mice (31), and therefore, a 4-week study was sufficient to elicit noticeable changes in %HbA1c. Both ELP-FGF21 and GLP1-ELP monotherapies significantly inhibited %HbA1c elevation relative to control, although chronic GLP1-ELP treatment was not sufficient to prevent a net of 1.0 ± 0.2% rise from days 0 to 27 (Fig. 4G and fig. S2B). The dual-agonist cohort exhibited the greatest degree of long-term glycemic control, with a minimal +0.3 ± 0.2% change in %HbA1c (P < 0.07 compared to GLP1-ELP) (Fig. 4G). Thus, treatment with the GLP1-ELP-FGF21 dual-agonist drug affords superior glycemic control compared to equimolar dosing of a long-acting GLP-1RA, likely through coordinated action of the GLP-1 and FGF21 components to enhance insulin secretion and increase insulin sensitivity, respectively.
Next, the metabolic effects of the GLP-1/FGF21 combination therapy were evaluated with respect to drug format. Db/db mice received a single subcutaneous injection of GLP1-ELP-FGF21 (1000 nmol/kg) or a mixture of each of GLP1-ELP (1000 nmol/kg) and ELP-FGF21 (1000 nmol/kg), and an ELP-only formulation (1000 nmol/kg) was included as a negative control. Body weights and ambient blood glucose levels were measured daily until the cohorts returned toward baseline, and an intraperitoneal GTT (0.75 mg/kg) was performed 6 days after treatment to evaluate how glycemic control was maintained over time.
Ambient blood glucose levels following GLP1-ELP-FGF21 treatment were reduced from >300 mg/dl and sustained at <150 mg/dl for ~10 days (Fig. 5A). These data confirm previously observed effects at the same dose (Fig. 3B) and are consistent with therapeutic drug levels maintained in the plasma out to ~10 days, as estimated from the dual-agonist pharmacokinetic profile (Fig. 3E). In contrast, the 1:1 mixture cohort maintained blood glucose at <150 mg/dl for only 4 days, after which the levels started returning to baseline (Fig. 5A). These results are consistent with the 1:1 mixture pharmacokinetic data, which show the plasma concentration of ELP-FGF21 dropping below the estimated minimal therapeutic level of 100 nm after day 4 (Fig. 4A), suggesting a crucial role for FGF21 in the combination treatment’s ability to maintain maximal glycemic control. AUC analysis of the ambient blood glucose versus time data confirmed that the GLP1-ELP-FGF21 treatment afforded significantly superior glucose control compared to the 1:1 mixture (Fig. 5B).

(A to F) Six-week-old db/db mice (n = 7) received a single subcutaneous injection of GLP1-ELP-FGF21 (1000 nmol/kg), a 1:1 equimolar mixture of GLP1-ELP and ELP-FGF21, or an ELP-only control. Ambient blood glucose levels were measured every 24 hours until animals returned to baseline levels and are reported as raw values (A) or blood glucose versus time AUC (B). (C to E) Glucose challenge. Six days after treatment, mice were fasted 5 hours, baseline blood glucose levels were measured (C), and mice were injected intraperitoneally with glucose (0.75 g/kg). Blood glucose levels were measured at indicated time points (D), and blood glucose versus time incremental AUC (iAUC) values were calculated (E). Body weights were recorded daily and are reported as a percentage change from preinjection weight over time (F). (G) Six-week-old db/db mice (n = 6 to 7) received a single subcutaneous injection of GLP1-ELP-FGF21 (1000 nmol/kg), a 1:1 equimolar mixture of GLP1-ELP and ELP-FGF21, or vehicle control. Food consumption was measured every 1 to 2 days and is reported as cumulative food intake per mouse. Data are presented as means ± SEM and were analyzed by one-way ANOVA or two-way repeated-measures ANOVA, followed by Dunnett’s tests. *P < 0.05 and ****P < 0.0001.
Fasting blood glucose levels measured before the GTT on day 6 were potently reduced upon GLP-1/FGF21 combination treatment and to an equivalent degree regardless of drug format (136.9 ± 9.6 mg/dl for GLP1-ELP-FGF21 and 129.3 ± 5.8 mg/dl for the GLP1-ELP + ELP-FGF21 mixture) (Fig. 5C). However, when compared to the day-6 ambient blood glucose levels (127.1 ± 4.7 mg/dl for GLP1-ELP-FGF21 and 232.4 ± 27.9 mg/dl for the GLP1-ELP + ELP-FGF21 mixture) (Fig. 5A), it is clear that glycemic control has started to wear off in the mixture-treated group at this time, whereas the dual-agonist cohort remains in the normoglycemic range regardless of fed versus fasted state. The ELP control displayed ambient and fasting glucose levels of 364.1 ± 29.0 and 324.1 ± 15.1 mg/dl, respectively, on day 6 (Fig. 5, A and C), demonstrating that the ELP alone, as expected, had no therapeutic activity.
While both the dual-agonist and the single-agonist mixture treatment cohorts displayed improved glucose tolerance (Fig. 5D), an incremental AUC (iAUC) analysis, which sets the baseline for each animal to its t = 0 blood glucose, revealed a significant reduction in the dual-agonist group only (Fig. 5E). Mice treated with GLP1-ELP-FGF21 recovered to near-baseline levels within 60 min (Fig. 5D), whereas the animal cohorts treated with the 1:1 mixture or the ELP did not return to their respective baselines following a glucose excursion. These results clearly demonstrate the robust degree of glucose tolerance that is exclusive to the dual-agonist formulation.
Both combination therapy formats induced weight loss—reducing body weights by 5.8 ± 0.8 and 6.5 ± 0.7% for the dual agonist and 1:1 mixture cohorts, respectively (Fig. 5F). GLP1-ELP-FGF21 inhibited weight gain to a greater degree, maintaining a net weight loss effect through day 8, while treatment with the 1:1 mixture maintained a net weight loss effect through only day 6 (Fig. 5F). These results further support the delivery of GLP-1 and FGF21 as a single molecule, as data point to a more consistent modulation of drug release into the systemic circulation. When food intake was tracked over 7 days following treatment, the dual-agonist and 1:1 mixture cohorts consumed chow at equivalent rates (Fig. 5G), indicating that differences in body weight gain were dependent on energy utilization and not to differential effects on satiety.
In summary, these findings clearly show that GLP-1 and FGF21 act synergistically in diabetic mice. Furthermore, fusion of GLP-1 and FGF21 to an ELP to create a depot-forming dual agonist results in a superior therapeutic outcome compared to a mixture of the two respective drugs, validating a multiagonist approach for the GLP-1/FGF21 combination therapy.